VCFdbR: A method for expressing biobank-scale Variant Call Format data in a SQLite database using R
Abstract
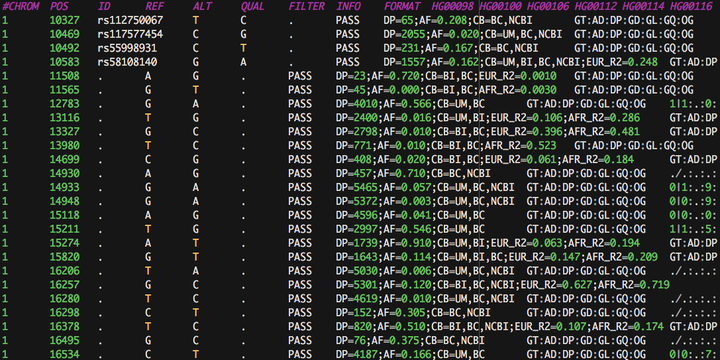
As exome and whole-genome sequencing cohorts grow in size, the data they produce strains the limits of current tools and data structures. The Variant Call Format (VCF) was originally created as part of the 1,000 Genomes project. Flexible and concise enough to describe the genetic variations of thousands of samples in a single flat file, the VCF has become the standard for communicating the results of large-scale sequencing experiments. Because of its static and text-based structure, VCFs remain cumbersome to parse and filter in an interactive way, even with the aid of indexing. Iterating on previous concepts, we propose here a pipeline for converting VCFs to simple SQLite databases, which allow for rapid searching and filtering of genetic variants while minimizing memory overhead. Code can be found at https://github.com/tkoomar/VCFdbR
Tanner Koomar, PhD
Postdoctoral Research Scholar
My research interests include computational genetics, machine learning, and science communication